Concetti importanti 7: Nomenclatura di idrocarburi saturi e insaturi, alcoli e ammine – Aggiornamenti IUPAC

Approfondimenti sulla nomenclatura IUPAC aggiornata su: IUPAC ‘Nomenclature of Organic Chemistry’ (Blue Book, 2013) e ‘Brief Guide to the Nomenclature of Organic Chemistry’ (IUPAC Technical Report, Pure Appl. Chem. 2020; 92(3): 527–539).
1. Introduzione e principi generali
La nomenclatura sostitutiva IUPAC costruisce nomi sistematici a partire da un composto genitore e dai gruppi sostituenti: il nome risultante combina la radice del genitore, suffissi che indicano insaturazioni o gruppi funzionali principali e prefissi che indicano sostituenti minori.
2. Regole per la scelta composto genitore (catena principale)
La scelta della catena principale è effettuata applicando la gerarchia di criteri IUPAC.
- La catena principale deve essere la catena con il maggior numero di atomi di carbonio, indipendentemente dalla presenza di insaturazioni.
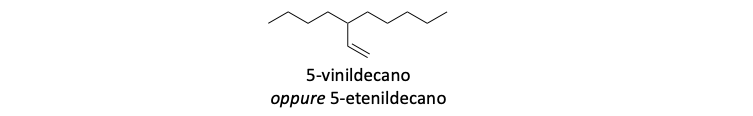
2. A parità di lunghezza si deve scegliere la catena più ramificata

3. A parità di lunghezza si deve scegliere la catena che presenta insaturazioni.
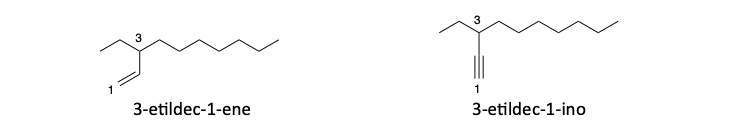
4. A parità di lunghezza si deve scegliere la catena con insaturazioni anche se non è la più ramificata

5. A parità di lunghezza si deve scegliere la catena con doppi legami rispetto a quella con tripli legami

6. A parità di lunghezza si deve scegliere la catena con il maggior numero di doppi legami, anche se non è la più ramificata

7. Se l’idrocarburo contiene un ciclo, questo deve esser considerato come composto genitore anche se ha un minor numero di carboni
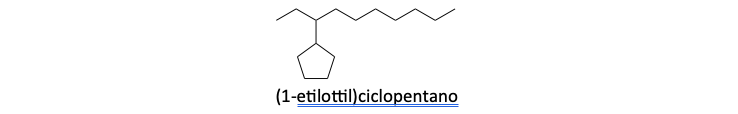
8. Il ciclo prevale anche se nella catena lineare c’è una insaturazione

9. In presenza di due cicli prevale quello con il maggior numero di carboni

10. Il ciclo con il maggior numero di carboni prevale anche se il ciclo più piccolo presenta un doppio legame

3. Nomenclatura dei gruppi alchilici, alchenilici e alchinilici
I gruppi alchilici, alchenilici e alchinilici derivano da un alcano, da un alchene o da un alchino, rispettivamente, per rimozione di un idrogeno. Il nome è formato dalla radice, che indica il numero di atomi di carboni, seguita dal suffisso ile; nel caso di sostituenti alchenilici e alchinilici, tra la radice e ile deve essere presente, rispettivamente, en o in.

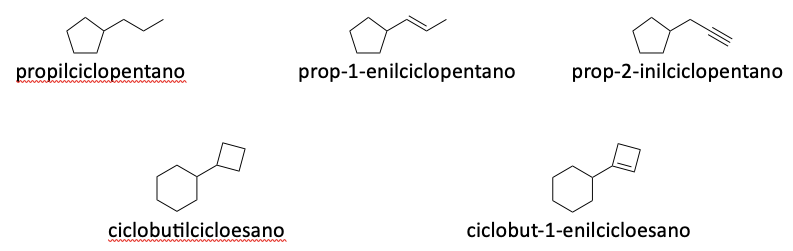
Se sul cicloalchenile è presente in altro sostuìituente, bisogna numerare il ciclo in modo da assegnare al doppio legame i numeri più bassi possibile.

4. Nomenclatura dei gruppi alchilidenici
Un alchilidene è un gruppo funzionale che si forma da un idrocarburo alifatico saturo per eliminazione di due atomi di idrogeno. Questi gruppi sono caratterizzati da un legame doppio tra il carbonio della catena principale e il gruppo alchilico derivato
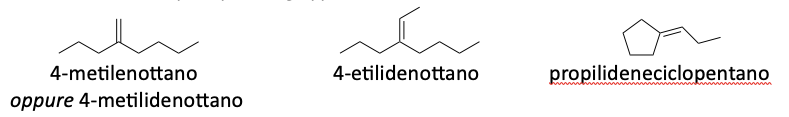
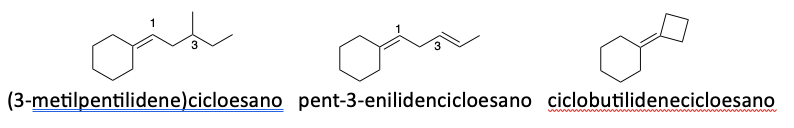
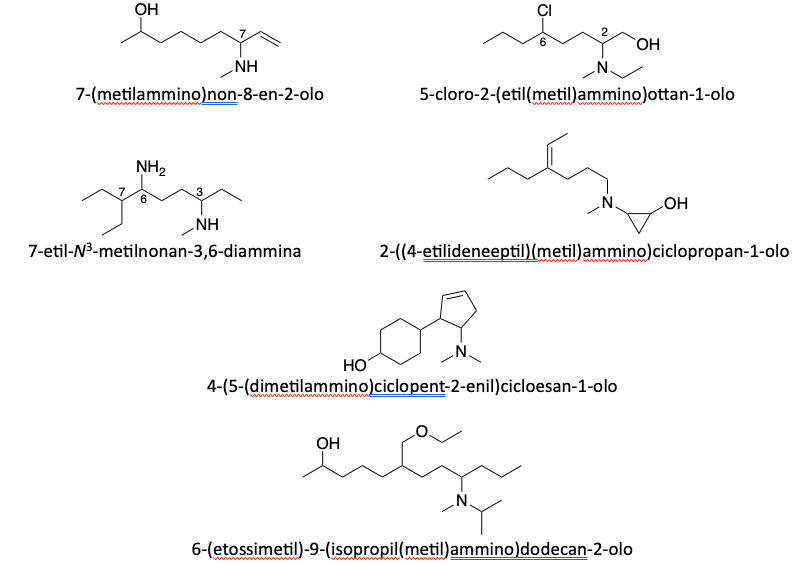
5. Nomenclatura di ammine e alcoli
Il gruppo alcolico e il gruppo amminico sono entrambi gruppi funzionali prioritari, ma il gruppo ossidrilico ha priorità superiore rispetto a quello amminico.
- Quando sono presenti in una molecola, la catena principale deve includerli, e al carbonio che li porta va assegnato il numero più basso possibile, anche se ciò comporta dare priorità minore alle insaturazioni.

2. Quando il gruppo ossidrilico o il gruppo amminico si trovano su un ciclo, il carbonio portante viene numerato come carbonio 1.
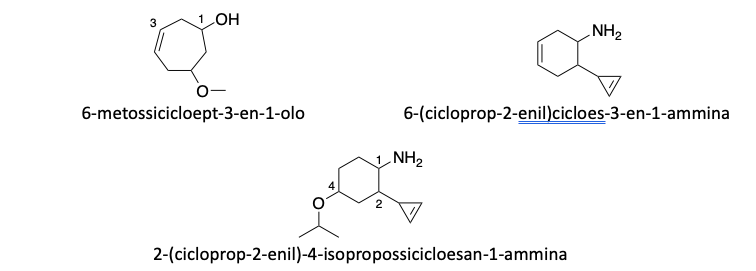
3. Quando entrambi i gruppi sono presenti nello stesso composto, il gruppo ossidrilico ha priorità e il composto genitore è considerato un alcol. Il gruppo amminico assume il ruolo di sostituente e viene indicato con il prefisso “ammino–”.

4. Quando i due gruppi si trovano su catene diverse della molecola, la catena che porta il gruppo ossidrilico viene considerata la catena principale

5. Quando due gruppi uguali (ossidrile o amminico) si trovano sulla stessa catena, prima del suffisso va aggiunto il prefisso “di–”.

6. Quando due gruppi uguali si trovano su catene diverse della molecola, quello presente sulla catena laterale deve essere indicato come sostituente.

7. Nel caso di ammine secondarie e terziarie, bisogna individuare la catena principale che porta l’azoto, seguendo le regole precedenti. Gli altri gruppi legati all’azoto vanno indicati come sostituenti, riportandone la posizione con la lettera N. Se lo stesso sostituente è presente sia sull’azoto sia sulla catena carboniosa, la N precede il numero.
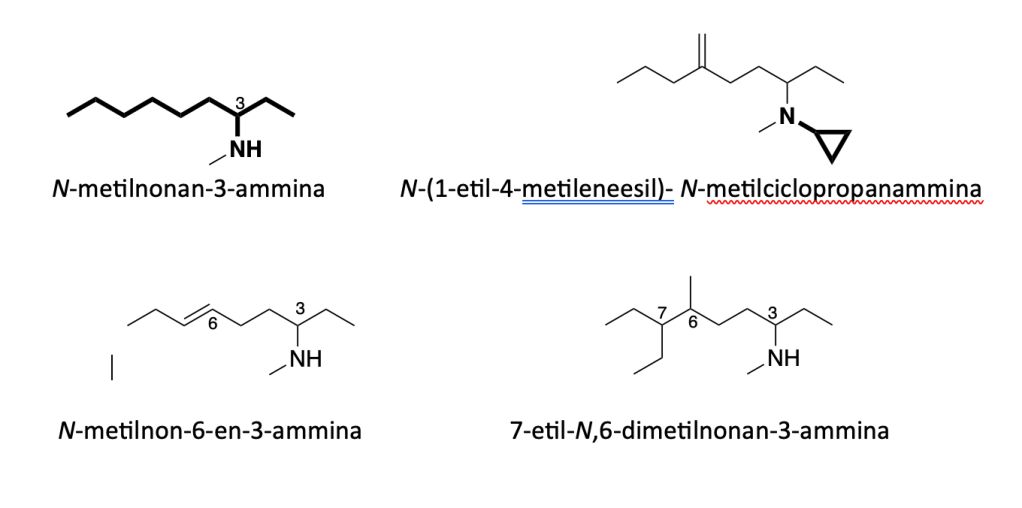
8. Nel caso in cui il gruppo amminico con uno o più gruppi alchilici è un sostituente, bisogna nominare i sostituenti, in ordine alfabetico, prima della parola “ammino”
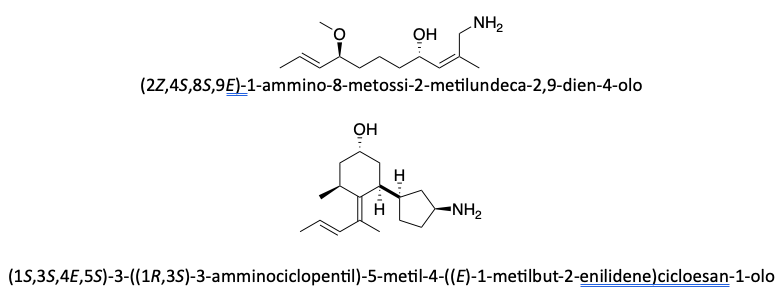
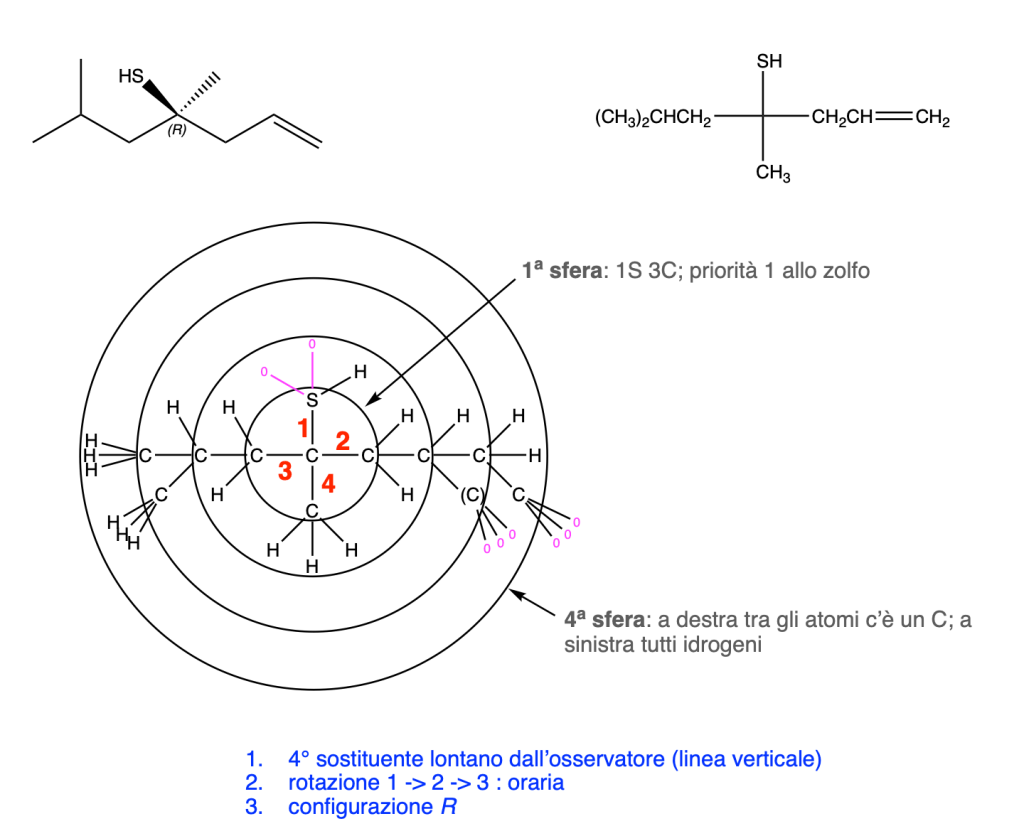
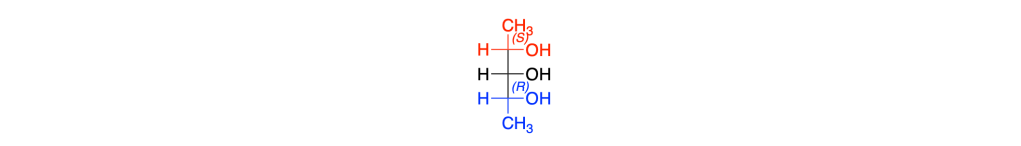
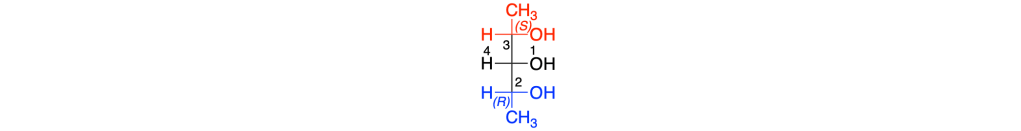
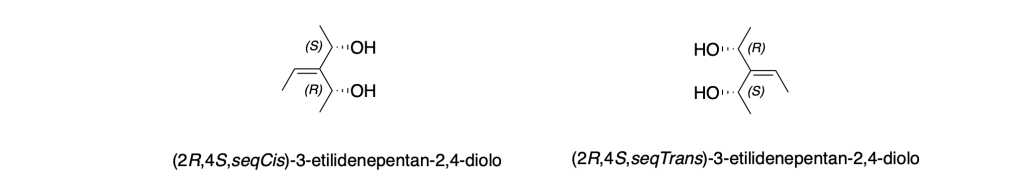
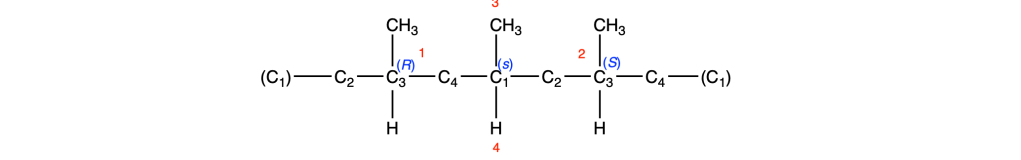
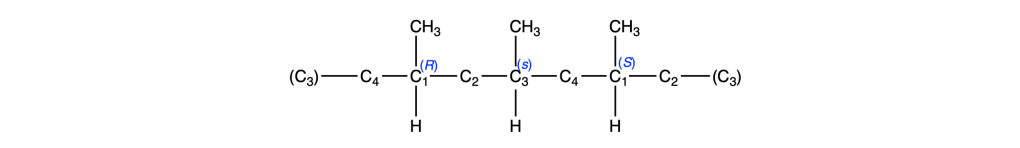
5. Stereochimica
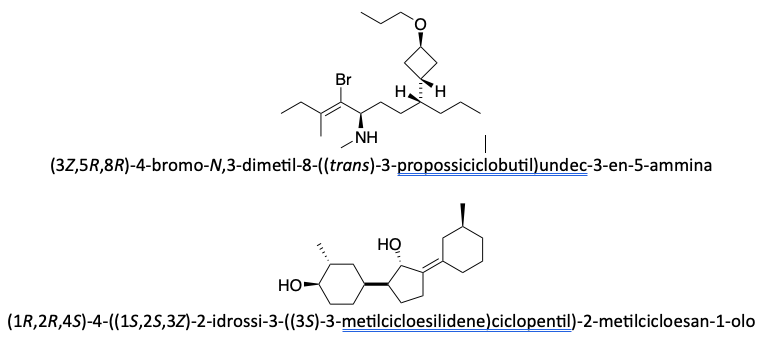
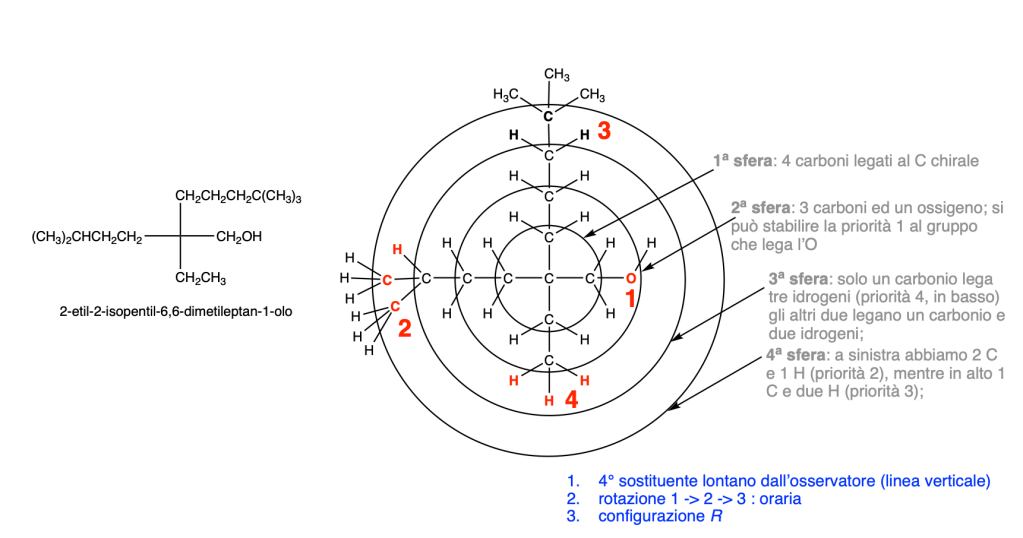
Prima del nome del composto va indicata la stereochimica tra parentesi, riportando la configurazione dei carboni chirali e dei doppi legami, seguendo l’ordine numerico delle loro posizioni. Le configurazioni dei sostituenti vanno specificate prima del nome dei sostituenti. Se sono presenti cicli disostituiti su carboni non chirali, l’orientazione dei sostituenti deve essere indicata con la notazione cis–trans.









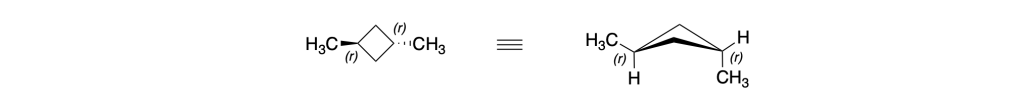

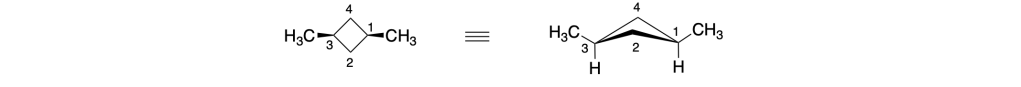

















You must be logged in to post a comment.