Concetti Importanti 17 – Cenni di stereochimica avanzata: Centri Pseudoasimmetrici ed Enantiomorfismo

Un centro pseudoasimmetrico è un atomo tetraedrico (di solito carbonio) che ha quattro sostituenti diversi, due dei quali sono enantiomeri tra loro, mentre gli altri due sono differenti da essi e tra loro. Questo centro è prochirale, ma non genera una molecola chirale nel suo complesso; piuttosto, si trova in una molecola achirale, anche se il centro ha una configurazione designata come r o s (in minuscolo), a differenza di R o S nei veri centri stereogenici.
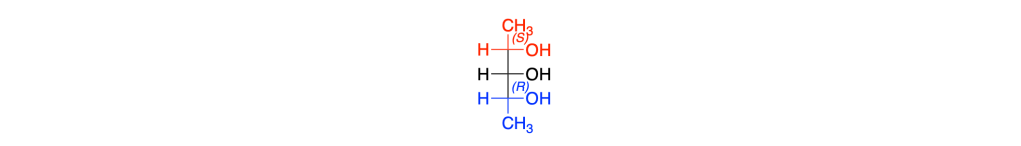
I sostituenti del carbonio 3 indicati in rosso e in blu sono gruppi chimici che sono immagini speculari tra loro e vengono detti sostituenti enantiomorfi
Il carbonio 3 del 2,3,4-pentantriolo (o molecole simili) non è asimmetrico quando il carbonio 2 e il carbonio 4 sono entrambi R (o entrambi S)
Quando uno dei due carboni è R e l’altro è S il carbonio 3 è asimmetrico; tale carbonio viene chiamato carbonio pseudoasimmetrico

La IUPAC raccomanda di usare r e s (minuscoli) per i centri pseudoasimmetrici, proprio per distinguerli dai veri centri stereogenici R/S
Per descrivere questi centri si utilizzano le regole CIP; poiché i due gruppi enantiomorfi sono costituiti dagli stessi atomi ma differiscono solo nella configurazione assoluta del carbonio chirale, il gruppo che ha il carbonio con configurazione R ha la priorità sul gruppo che ha il carbonio con configurazione S. Pertanto, nel caso del primo composto le priorità sono le seguenti:
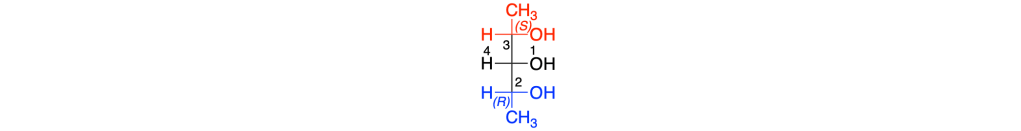
La rotazione 1=> 2 =>3 è oraria ma il quarto sostituente si trova sulla linea orizzontale della proiezione di Fischer, la notazione del carbonio 3 sarà s.
Il nome IUPAC della prima forma meso sarà dunque (2R, 3s,4S)-2,3,4-pentantriolo; la seconda forma meso avrà il nome (2R, 3r,4S)-2,3,4-pentantriolo
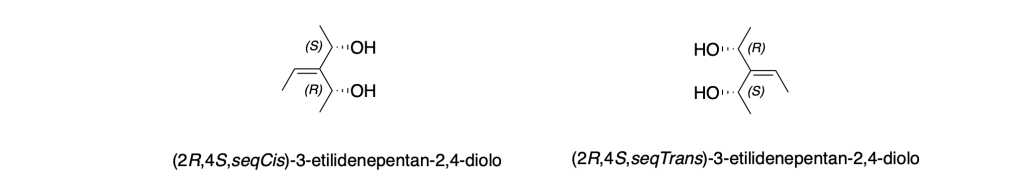
Si definisce enantiomorfo un doppio legame in cui i gruppi legati creano due configurazioni speculari non sovrapponibili, a causa della presenza di ligandi chirali.
I gruppi chirali presenti sulle due estremità sono enantiomorfi, ovvero configurati in modo opposto (es. un (R) da un lato e un (S) dall’altro).
In tal caso, la configurazione del doppio legame (che normalmente sarebbe descritta come cis/trans o Z/E) non è sufficiente a distinguere le due forme, perché quelle due configurazioni possono essere enantiomorfe fra loro.
In questi casi, la IUPAC ha introdotto i descrittori seqCis e seqTrans:
• seqCis: i due sostituenti con priorità più alta secondo le regole CIP sono dallo stesso lato.
• seqTrans: si trovano su lati opposti.
Questi descrittori sono riflessioni l’uno dell’altro, e quindi rappresentano una coppia di enantiomorfi geometrici.
Anche i carboni 1 e 3 del trans-1,3-dimetilciclobutano sono carboni pseudoasimmetrici:
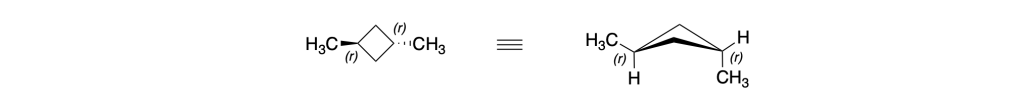
Infatti, anche per questo composto, la IUPAC suggerisce il nome (1r,3r)-1,3-dimetilciclobutano piuttosto che del trans-1,3-dimetilciclobutano.
Analogamente, il nome preferito dalla IUPAC per il cis-1,3-dimetilciclobutano è (1s,3s)-1,3-dimetilciclobutano.

Come si fa in questo caso ad assegnare la notazione r o s?
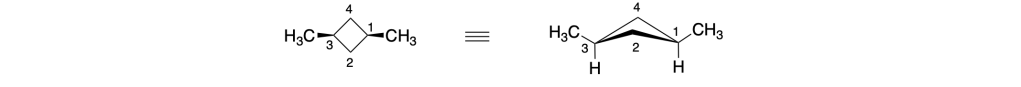
Immaginiamo di metterci nel centro dell’anello e di guardare il carbonio 1; riportiamo questo carbonio in una proiezione di Fischer: se il carbonio 1 si trova al centro della croce, in alto avremo il gruppo metile, in basso l’idrogeno a destra il C2 e a sinistra il C4; Continuando a destra, dopo il C2 abbiamo il C3 che lega il metile in alto e l’idrogeno in basso, poi il C4 e infine ritorniamo a C1: l’ultimo termine della sequenza è un carbonio duplicato (C1) che si intende legato a tre atomi fantasma

Il carbonio 3, è chirale in questa rappresentazione, pertanto possiamo assegnare le priorità ai gruppi legati sulla base delle regole CIP: l’H è l’atomo con priorità più bassa (4° sostituente) e il metile è il sostituente 3. Il C2 lega due idrogeni e un carbonio così come il C4, pertanto dobbiamo andare oltre. Il C2 è legato ad un carbonio che lega un idrogeno e due carboni, mentre il C1 è rappresentato come legato a tre atomi fantasma con numero atomico nullo (0). Quindi il gruppo a sinistra ha priorità 1 mentre il gruppo legato a destra, priorità 2. La rotazione è antioraria, il quarto sostituente sulla linea verticale, pertanto la configurazione assoluta è S. Facendo lo stesso a sinistra e assegnando le priorità troviamo che il C3 a sinistra ha una configurazione R:

Possiamo notare che i gruppi legati a C1 sul lato destro e sinistro dell’anello sono enantiomorfi, pertanto C1 è un centro pseudoasimmetrico. Assegnando le priorità e ricordando che il sostituente enantiomorfo con configurazione R ha la priorità sul sostituente enantiomorfo con configurazione S, assegniamo a C1 la notazione s.
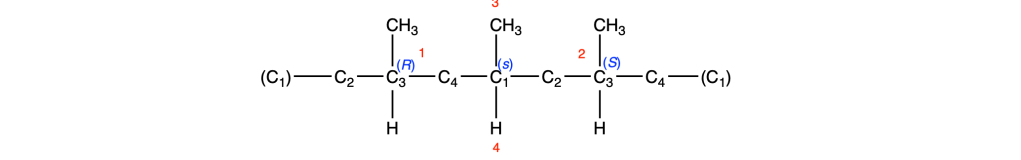
Facendo la stessa cosa per il C3, troviamo una situazione identica:
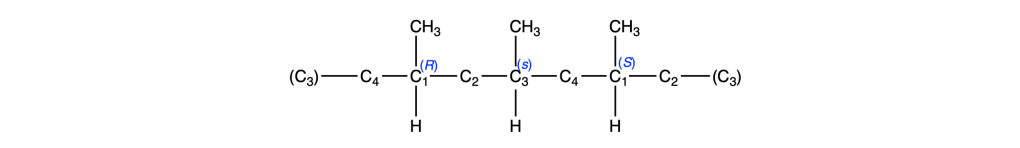
Da cui ricaviamo il nome IUPAC del composto come (1s,3s)-1,3-dimetilciclobutano.
Quando un carbonio lega quattro sostituenti diversi ma due sono gruppi alchenilici che che si distinguono unicamente per la configurazione dei doppi legami, il carbonio è chirale e nell’assegnare la configurazione assoluta, il doppio legame con configurazione Z ha la priorità sul doppio legame con configurazione E.




















































































You must be logged in to post a comment.