Concetti importanti 12: Nomenclatura dei composti polifunzionali

Se una molecola organica contiene due o più gruppi funzionali, dobbiamo scegliere solo un suffisso per attribuire il nome; non è corretto utilizzare due o più suffissi.
As esempio l’amminoalcol 1 deve essere nominato come un alcol con il suffisso -olo o come un’ammina con il suffisso -ammina ma non può avere il suffisso -oloammina o -amminolo.
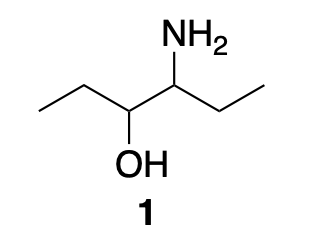
L’unica eccezione a questa regola è quando bisogna dare un nome si un composto che ha un doppio e/o un triplo legame: in questo caso si utilizza il nome alchenino dando alle insaturazioni il numero più basso. In caso di parità la priorità spetta al doppio legame.

L’alcol insaturo si chiamerà ott-4-en-2-olo, mentre l’ammina insatura ott-7-in-2-ammina:

Come scegliamo quale suffisso utilizzare? I gruppi funzionali sono divisi in due classi, gruppi principali e gruppi subordinati, come mostrato nella tabella seguente. I gruppi principali possono essere citati sia come prefissi sia come suffissi, mentre i gruppi subordinati sono citati solo come prefissi. All’interno dei gruppi principali, è stata stabilita un ordine di priorità: il suffisso corretto per una data molecola è determinato scegliendo il gruppo principale di massima priorità. Ad esempio, la tabella seguente indica che l’amminoalcol 1 dovrebbe essere nominato come un alcol piuttosto che come ammina perché un gruppo funzionale dell’alcol è di maggiore priorità rispetto a un’ammina.
Composti organici elencati in base all’ordine di priorità dei gruppi funzionali
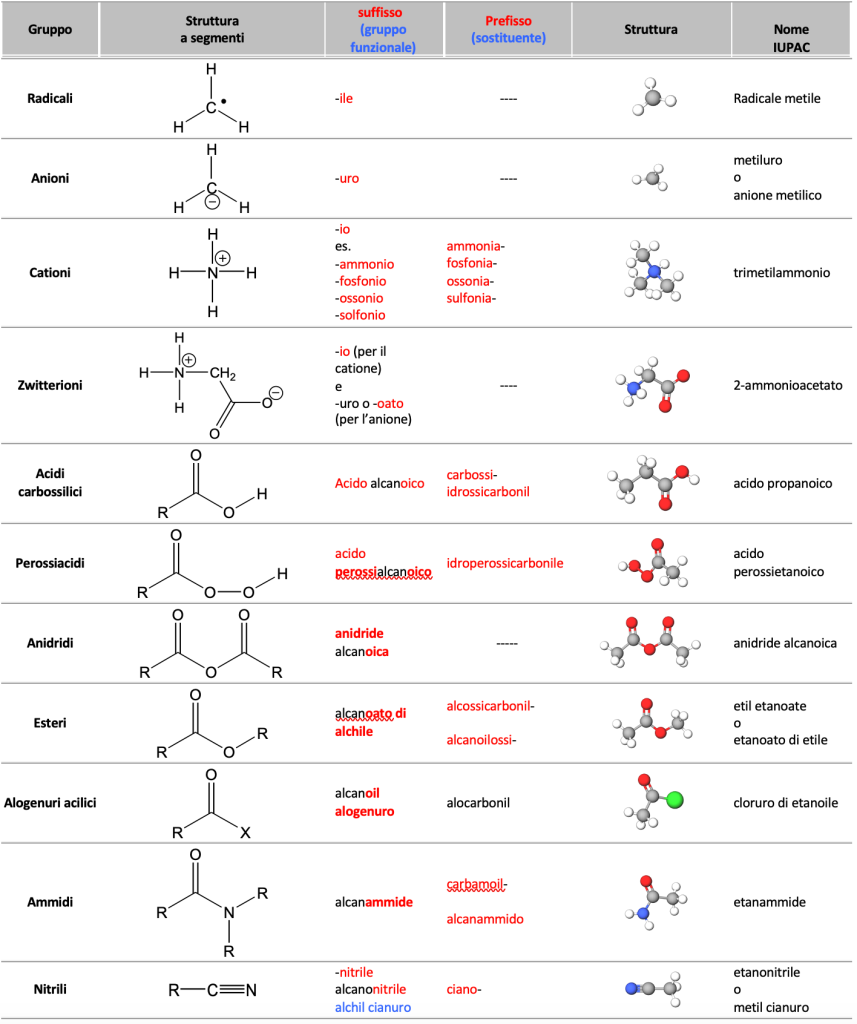
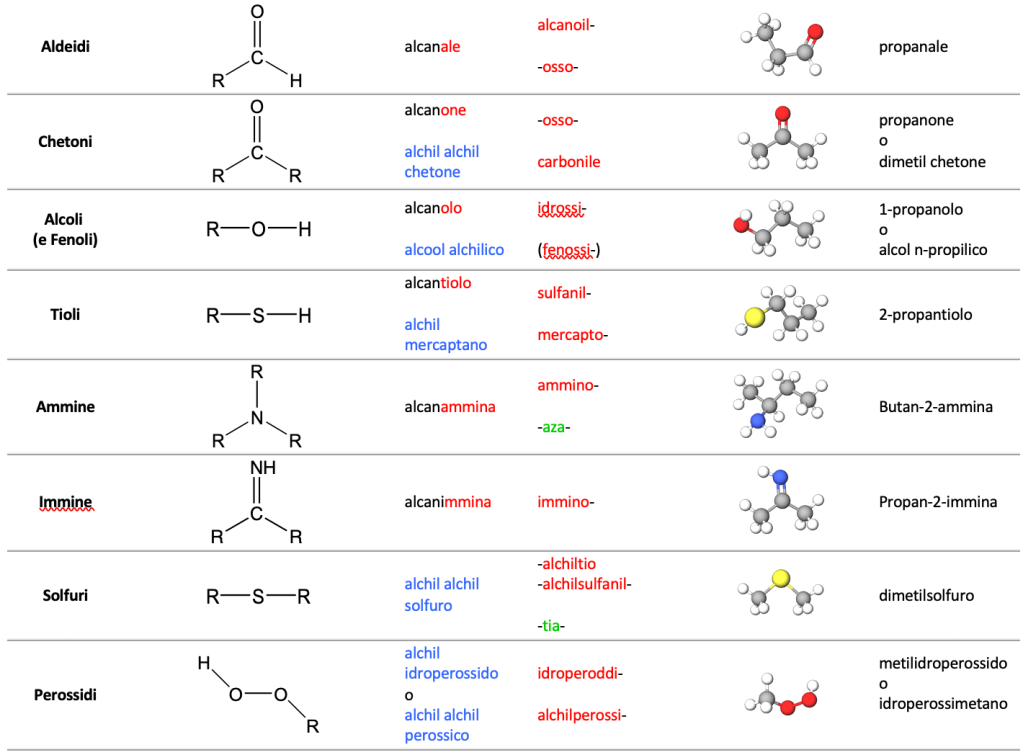

Il nome parentale, o base, di un composto organico polifunzionale è di solito facile da identificare. Se il gruppo prioritario fa parte di una catena aciclica, il nome base è quello della catena più lunga che contiene il gruppo prioritario, indipendentemente dalla presenza di altri gruppi funzionali o di insaturazioni; se il gruppo principale prioritario si trova su un ciclo, sarà il ciclo ad avere il nome base e le catene lineari, anche se più lunghe, saranno nominate come sostituenti.

Ricorda che a parità di gruppo funzionale, il ciclo ha sempre la priorità, indipendentemente dal numero di carboni della catena lineare o dalla presenza di insaturazioni o di altri gruppi funzionali.

Con il nome base e il suffisso stabilito, il passo successivo è identificare e assegnare numeri a tutti i sostituenti sulla catena principale o sull’anello. I sostituenti includono tutti i gruppi alchilici e tutti i gruppi funzionali diversi da quello citato nel suffisso.
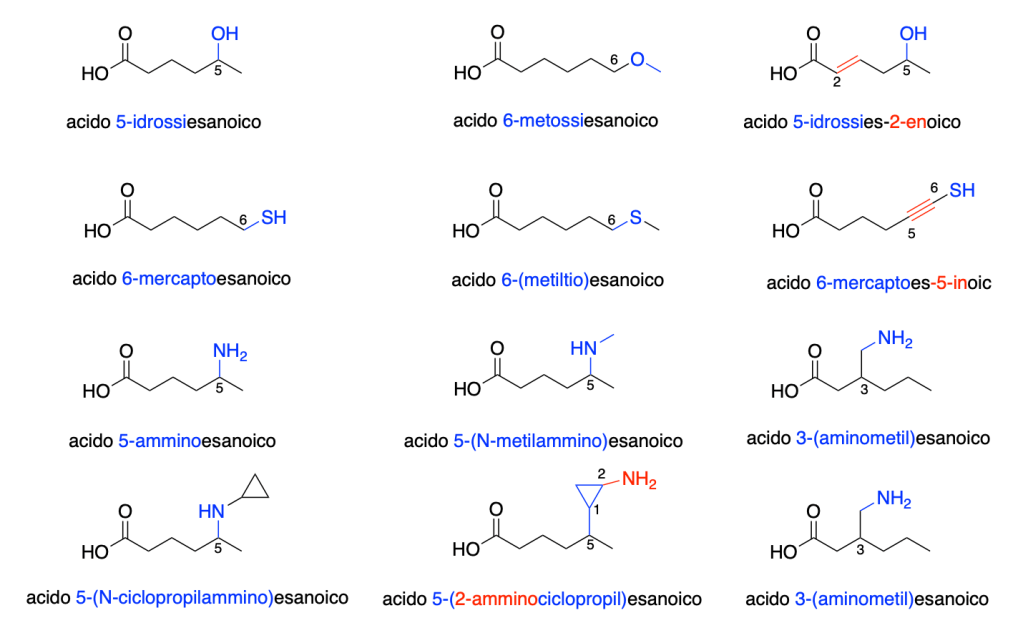
Ognuno di questi acidi contiene più gruppi funzionali. Poiché il gruppo carbossile è prioritario, la catena più lunga deve contenre il gruppo carbossilico; inoltre, bisogna numerare dall’estremità più vicina al gruppo funzionale prioritario.
Nel caso di acidi carbossilici contenenti altri gruppi carbonilici, il gruppo carbossilico è sempre il gruppo prioritario
Derivati di acidi carbossilici come sostituenti:

Esempi:
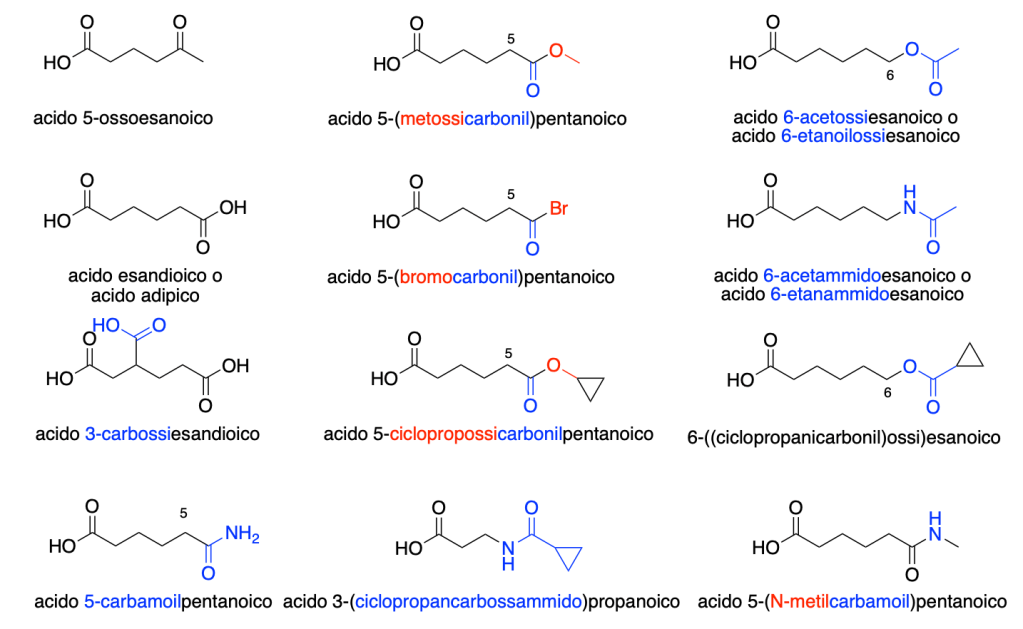
Esempi di acidi carbossilici contenenti cicli:

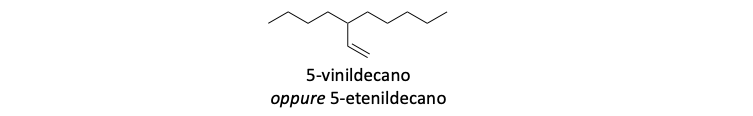
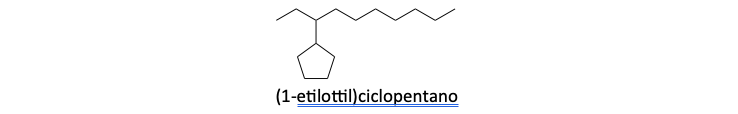
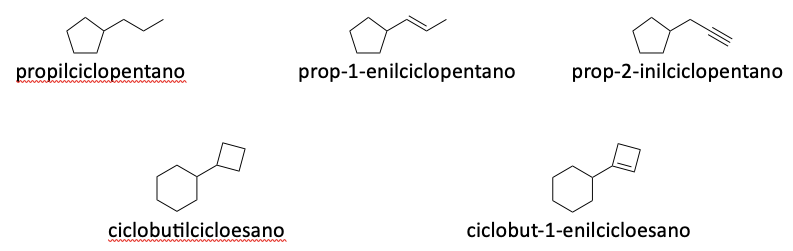
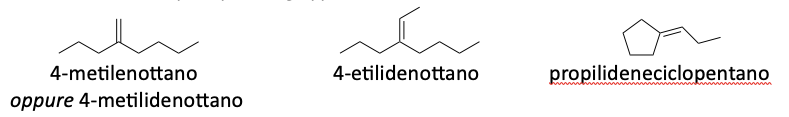
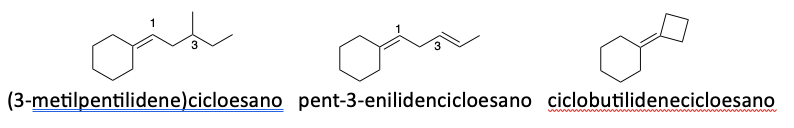
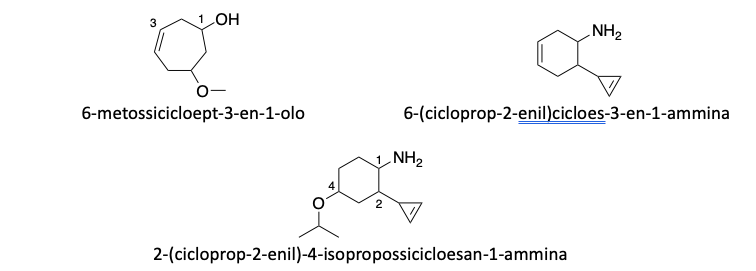
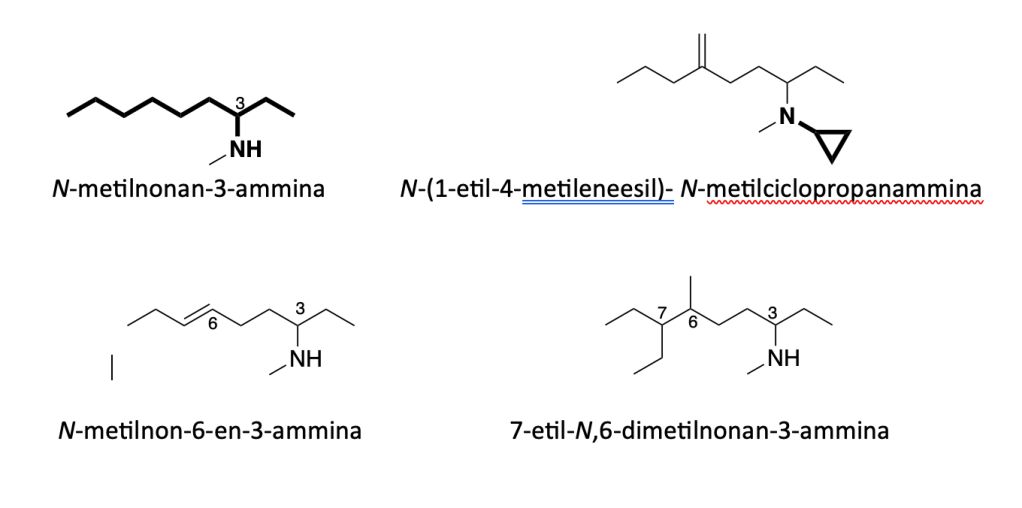
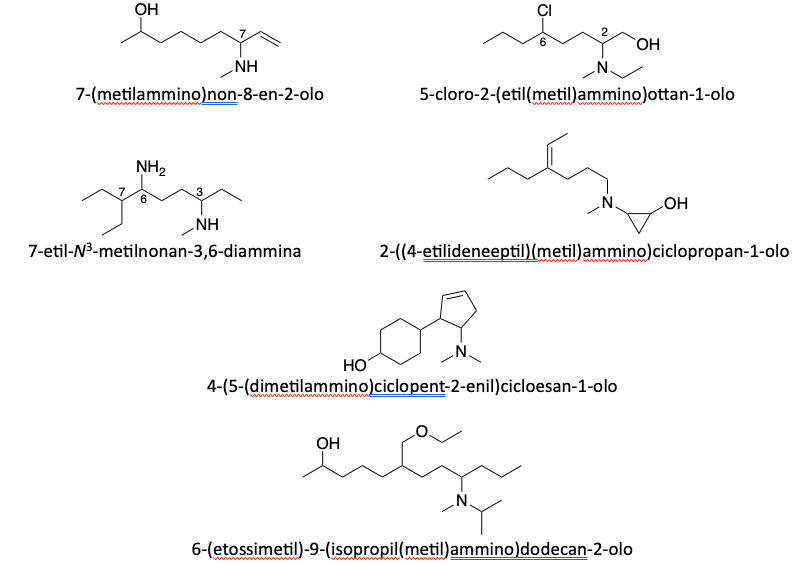
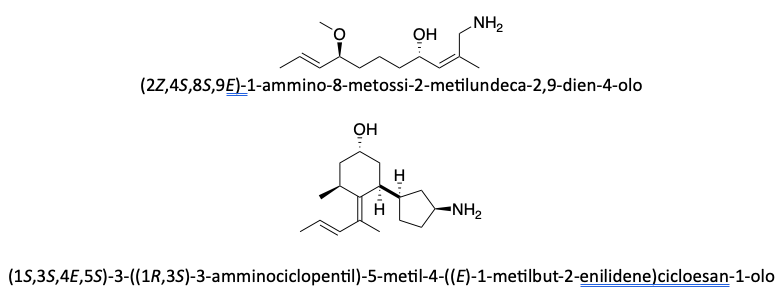
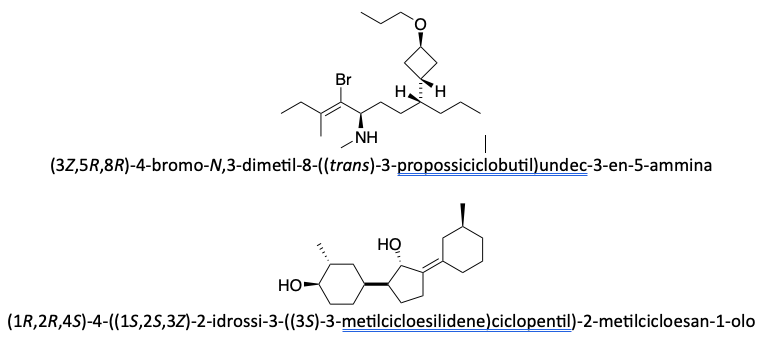
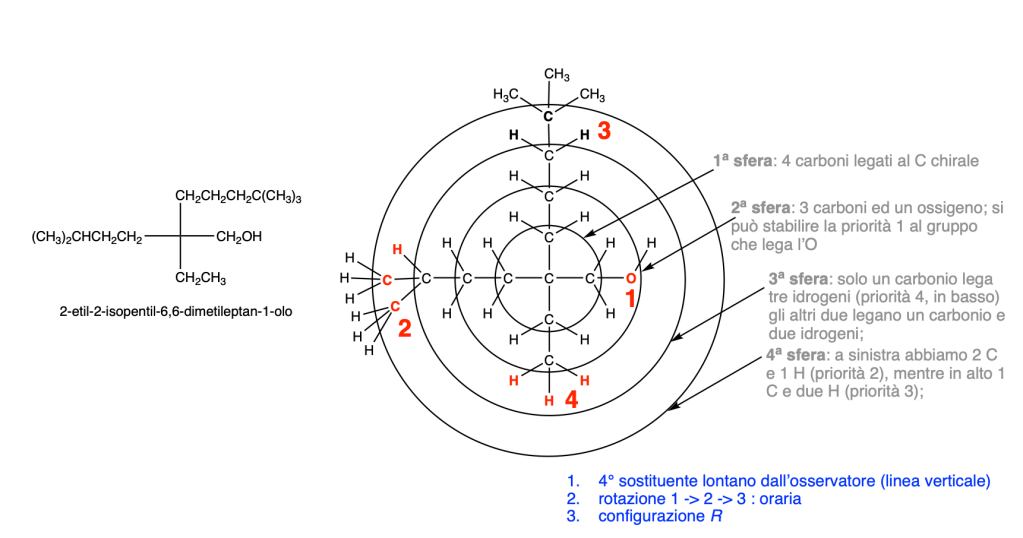
Altri esempi di nomenclatura di composti polifunzionali:

































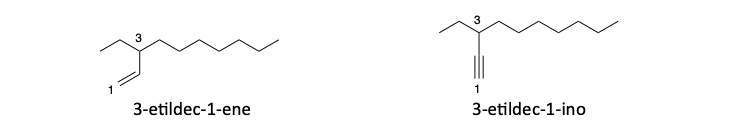
































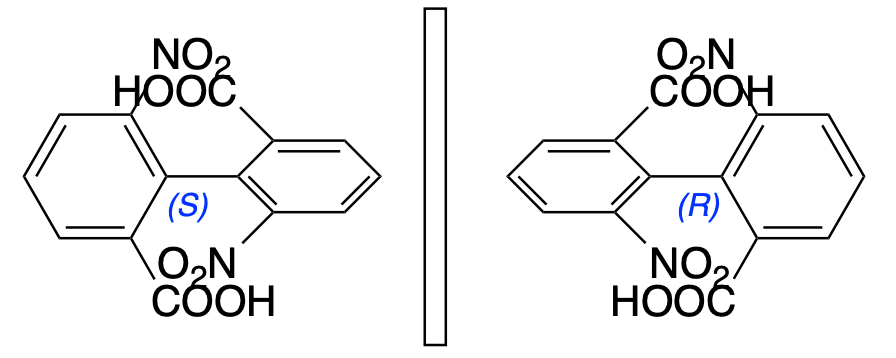
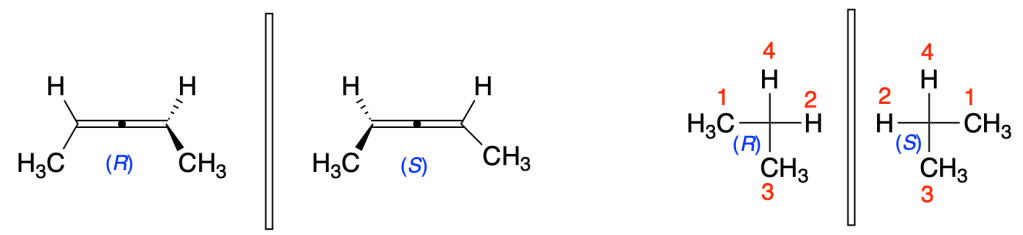



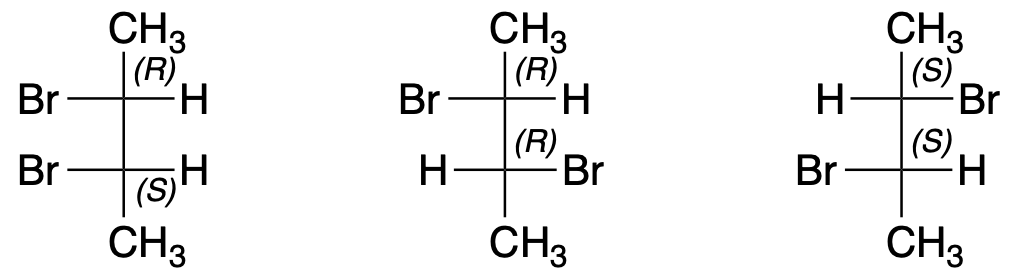
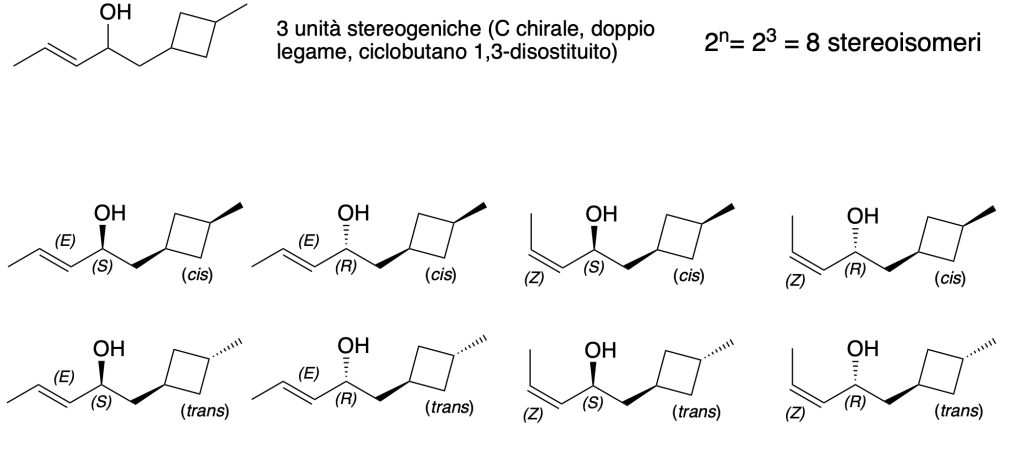




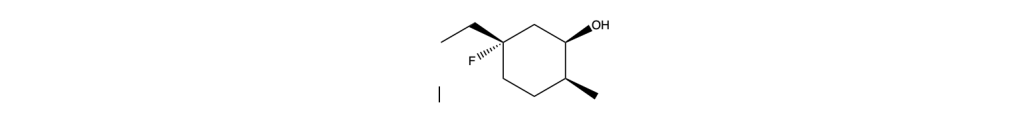
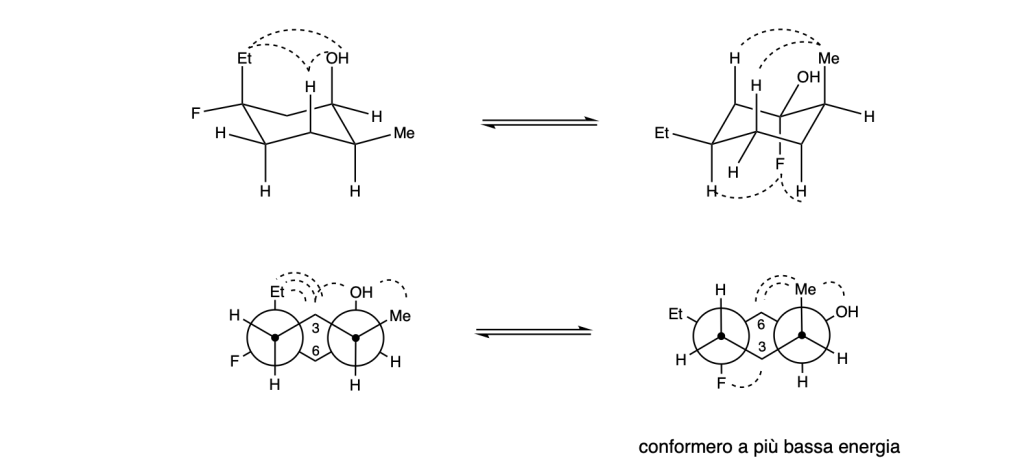


You must be logged in to post a comment.