Nel testo di riferimento (il Bruice) alcuni meccanismi non sono esplicitati (o in alcuni casi non lo sono per esteso), data la somiglianza con altri meccanismi (o per il fatto che si tratta di meccanismi già spiegati altrove).
Qui troverete un elenco di questi meccanismi con indicazioni su dove trovarli o con spiegazioni relative alla loro estrapolazione da meccanismi già presenti sul libro:
Sintesi di Gabriel: Dopo aver sintetizzato l’immide N-sostituita, questa deve essere idrolizzata. Ci sono diversi metodi per ottenere questa idrolisi, quello indicato sul testo è l’idrolisi in ambiente acido. Il meccanismo ricorda quello dell’idrolisi di un’ammide catalizzata da acidi (la cui spiegazione potete utilizzare a supporto della comprensione del meccanismo qui riportato; NB: per visualizzare i commenti ai singoli passaggi, è necessario scaricare il file pdf).
Transesterificazione catalizzata da acidi: meccanismo identico all’idrolisi dell’estere catalizzata da acidi. Il meccanismo per esteso è presente nel file delle correzioni degli esercizi su Reazioni di sostituzione nucleofila acilica (Ex 1a). Per la spiegazione relativa ad ogni passaggio, far riferimento al meccanismo di idrolisi dell’estere catalizzata da acidi.
Transesterificazione favorita da base: meccanismo identico all’idrolisi dell’estere favorita da ione idrossido. Il meccanismo per esteso è presente nel file delle correzioni degli esercizi su Reazioni di sostituzione nucleofila acilica (Ex 1e). Per la spiegazione relativa ad ogni passaggio, far riferimento al meccanismo di idrolisi dell’estere favorita da ioni idrossido.
Esterificazione di Fischer: il meccanismo è l’esatto contrario del meccanismo di idrolisi dell’estere catalizzata da acidi. Esso è presente nel file delle correzioni degli esercizi su Reazioni di sostituzione nucleofila acilica (Ex 4c). Per la spiegazione relativa ad ogni passaggio, far riferimento al meccanismo di idrolisi dell’estere catalizzata da acidi (ovviamente tenendo conto che si tratta del meccanismo inverso).
Idrolisi di un nitrile catalizzata da acidi: il meccanismo è riportato sul libro, ma ad un certo punto, dopo la formazione dell’ammide protonata, si fa riferimento ad “alcuni stadi”. Questi stadi non sono altro che quelli descritti nel meccanismo di idrolisi di un’ammide catalizzata da acidi (pag. 712, a partire dal secondo passaggio).
Idrolisi immina: il meccanismo corrisponde alla reazione inversa rispetto alla sintesi dell’immina (ma fare attenzione alla irreversibilità dell’idrolisi, a causa delle condizioni di reazione–> vedi spiegazione a pag. 762) e sarà esplicitato nel file relativo alle correzioni degli esercizi del post relativo alle Biomolecole (correzioni che non sono state ancora caricate)
Idrolisi emammine: scaricare il file qui. Valgono le stesse considerazioni fatte per l’idrolisi dell’immina.
Idrolisi acetale: il meccanismo corrisponde alla reazione inversa rispetto alla sintesi dell’acetale ed è esplicitato sul libro nella “strategia per la risoluzione dei problemi” a pag. 771.
Meccanismo di ciclizzazione degli zuccheri: sarà riportato nel file relativo alla correzione degli esercizi sulle Biomolecole. NB: le correzioni saranno rese disponibili entro il prossimo fine settimana.
Sintesi di Kiliani-Fischer: Si tratta di reazioni già note, in ogni caso i meccanismi per esteso saranno riportati nel file relativo alla correzione degli esercizi sulle Biomolecole. Per le spiegazioni è possibile far riferimento ai paragrafi del libro in cui sono spiegati i singoli meccanismi.
Sintesi dei peptidi: Pur essendo i meccanismi coinvolti riportati sul libro, alcuni passaggi sono sottintesi. Un meccanismo più dettagliato sarà riportato nel file relativo alla correzione degli esercizi sulle Biomolecole. Da integrare con le spiegazioni presenti sul testo. Per l’ultimo passaggio (idrolisi con acido trifluoroacetico) non è richiesto il meccanismo.
Utilizzandoquesto form è possibile segnalare eventuali meccanismi per cui non si ha ancora a disposizione materiale sufficiente. NB: Consultare attentamente il libro e il materiale caricato sul blog (incluse le correzioni degli esercizi) PRIMA dell’eventuale compilazione del form, che si chiuderà il 16/01.
Con lo studio della nomenclatura dei composti carbonilici, è arrivato il momento di mettere insieme tutte le regole studiate fino ad ora per poter essere in grado di attribuire il nome IUPAC a molecole anche complesse. Nello schema qui riportato, bisogna modificare la prima domanda in “il composto contiene un gruppo funzionale prioritario”?
Sarà il gruppo funzionale prioritario a determinare lo scheletro principale e il verso di numerazione.
Ai fini della nomenclatura, una volta individuato il gruppo funzionale prioritario, tutti gli altri gruppi funzionali vanno considerati alla stregua di sostituenti e vanno indicati come prefissi (fanno eccezione i legami multipli).
NB: in un ciclo, il gruppo funzionale prioritario sarà sul carbonio 1. La numerazione procede nel verso che dà all’eventuale legame multiplo il numero più basso; se non ci sono legami multipli, si procede nella direzione che dà al primo sostituente il numero più basso. Su una catena lineare, invece, si deve partire da una delle estremità e si sceglierà la direzione che dà al carbonio che lega il gruppo funzionale prioritario il numero più basso possibile.
Nella dispensa che è possibile scaricare utilizzando il link precedente troverete anche indicazioni su come indicare la presenza di gruppi carbonilici come sostituenti (vedere paragrafo Prefissi da usare quando è presente un gruppo funzionale terminale non prioritario).
NB: troverete qui riferimenti a gruppi funzionali che non abbiamo ancora studiato…per ora concentratevi su quelli già introdotti a lezione!
ATTENZIONE: Non tutte le regole qui riportate sono aggiornate in accordo con le indicazioni IUPAC 2013, dato che le regole utilizzate prima dell’ultima revisione sono ancora ampiamente utilizzate.
Tutti gli aggiornamenti già introdotti possono essere consultati ai link seguenti:
Le reazioni che avvengono a carico di questi composti, non sono altro che reazioni di sostituzione ed eliminazione, che qui andremo a schematizzare. Per ora abbiamo approfondito solo quelle che avvengono a carico degli alcoli. Gli altri meccanismi saranno approfonditi giovedì a lezione.
ALCOLI Gli alcoli possono essere convertiti in alogenuri alchilici (i quali possono poi essere convertiti in una grande varietà di composti, in virtù della loro reattività) o alcheni. La prima trasformazione avviene mediante reazioni di sostituzione, la seconda mediante una reazione di eliminazione (dato che viene eliminata una molecola di acqua, si parla di disidratazione).
Le reazioni da conoscere sono le seguenti:
In blu sono riportate le reazioni di sostituzione, in nero quelle di disidratazione. HX= HBr, HI, o HCl/ZnCl2
Il meccanismo con cui le reazioni avvengono, dipende dalla natura dell’alcol (primario, secondario o terziario):
Per quanto riguarda gli aspetti stereochimici, valgono le stesse considerazioni fatte per le reazioni che avvengono a carico degli alogenuri alchilici.
ETERI Gli eteridanno reazioni di sostituzione nucleofila con HBr e HI (la reazione è lenta, per cui la miscela di reazione deve essere riscaldata). Il meccanismo sarà SN1, se, dopo la protonazione del gruppo OR, allontanandosi questo dà luogo ad un carbocatione stabile. Se questo non accade, il meccanismo sarà di tipo SN2.
EPOSSIDI Gli epossidi possono reagire con un gran numero di nucleofili e il meccanismo di apertura dell’epossido dipende dalle condizioni in cui avviene la reazione:
La reazione di apertura dell’epossido avviene con attacco da retro, determinando la stereochimica del prodotto: l’ossidrile (che deriverà dall’ossigeno dell’epossido) e il nucleofilo (che ha dato l’attacco) si troveranno in anti tra loro.
Tantissimi nucleofili possono reagire con gli epossidi, tra questi, ad esempio, gli ioni acetiluro (portando ad un allungamento della catena carboniosa), ione idrossido (che porta alla sintesi di dioli trans), idruri (sintesi di alcoli senza trasposizione).
Per i dettagli teorico-pratici dei singoli meccanismi, si rimanda al testo
Una reazione SN2 è bimolecolare: sia l’alogenuro alchilico sia il nucleofilo sono coinvolti nello stato cineticamente determinante della reazione, per cui la velocità della reazione dipende dalla concentrazione di entrambi. Nel video seguente, è possibile seguire la reazione:
Nella seguente immagine, sono mostrati gli orbitali coinvolti:
Una reazione SN1 è unimolecolare: solo l’alogenuro alchilico è coinvolto nello stato di transizione dello stadio lento della reazione, per cui la velocità dipende solo dalla sua concentrazione. Nel video seguente, è possibile seguire la reazione:
Questa reazione prevede nel primo passaggio la formazione di un carbocatione, successivamente attaccato da un nucleofilo. Il carbonio carbocationico sarà ibridato sp2, per cui sarà l’orbitale p vuoto ad accettare gli elettroni del nucleofilo. In ChemTube 3D, è possibile visualizzare gli orbitali coivolti (valgono le stesse indicazioni di sopra, ma qui bisogna guardare sia lo step 1 sia lo step 2).
E2
L’E2 è una reazione bimolecolare, concertata, ad uno stadio, in cui il protone e lo ione alogenuro vengono rimossi contemporaneamente. La reazione E2 avviene solo se il legame con l’idrogeno e con il gruppo uscente sono paralleli (dato che i due orbitali sp3 dovranno diventare gli orbitali p che si sovrapporranno per formare l’orbitale pi greco) ed in particolare è favorita se i due gruppi si trovano in una conformazioneantiperiplanare. L’eliminazione anti è favorita perché il conformero che reagisce è un conformero sfalsato, inoltre, in questo modo si evita la repulsione tra la base e il gruppo uscente e, infine, nel caso dell’eliminazione anti, si ha la sovrapposizione ottimale degli orbitali, come mostrato nella seguente animazione:
In una reazione E2 i due gruppi eliminati da un anello a sei termini devono essere entrambi in posizione assiale (solo in quel caso sono antiperiplanari). La reazione E2 è stereoselettiva. Inoltre, il fatto che i due gruppi da eliminare si debbano trovare in posizione antiperiplanare, determina anche la stereospecificità della reazione qualora il carbonio beta sia legato ad un solo idrogeno.
E1
Una reazione E1, unimolecolare, è una reazione in due stadi in cui l’alogenuro alchilico si dissocia formando un carbocatione intermedio. Poi, una base rimuove un protone da un carbonio adiacente al carbonio carico positivamente, come mostrato nel seguente video:
Competizione tra i diversi meccanismi
Gli alogenuri alchilici vanno incontro a questo tipo di reazioni:
…e il prevalere di un meccanismo sull’altro dipende dalle caratteristiche strutturali dell’alogenuro alchilico, ma anche dal tipo di nucleofilo/base e, spesso, dalle condizioni di reazione. Al link seguente (vedere testo in rosso) potrete trovare uno schema e una tabella che, insieme alla conoscenza di tutti i fattori determinanti, può aiutarvi a capire con quale meccanismo avverrà una reazione o supportarvi nella scelta di una determinata reazione in una strategia sintetica: REAZIONI DI SOSTITUZIONE ED ELIMINAZIONE
NB: questi schemi non si applicano agli alogenuri benzilici e allilici (vedere libro).
Solventi
Un’ultima nota è relativa ai solventi utilizzati nelle reazioni.. Per quanto riguarda gli effetti sulle reazioni, si rimanda al libro, ma a questo punto è opportuno fare un elenco dei solventi di impiego più comune. Questi possono essere distinti in apolari e polari. Questi ultimi, sono distinti in polari protici e aprotici. I solventi apolari hanno momento dipolare nullo o molto piccolo. Esempi: esano, cloroformio, etere etilico, benzene. Alcuni solventi caratterizzati da una moderata polarità e che sono aprotici sono diclorometano, tetraidrofurano (THF) e acetato di etile. Esempi di solventi aprotici polari sono l’acetone, l’acetonitrile, il dimetilsolfossido (DMSO), la dimetilformammide (DMF). I solventi protici hanno un idrogeno legato ad un atomo di ossigeno oppure di azoto; ne sono esempi l’acqua, il metanolo, l’etanolo, il butanolo, l’acido acetico, l’ammoniaca. Da notare come la struttura chimica determini le caratteristiche dei solventi.
Tabelle dei solventi di comune impiego in chimica organica, con le loro caratteristiche chimico-fisiche, possono essere consultate ai seguenti link:
Nel materiale qui condiviso, sono compresi anche aspetti che non sono stati affrontati a lezione, inclusi degli approfondimenti su aspetti che dovreste già conoscere (ad esempio la relazione tra l’energia di un fotone e la frequenza della radiazione elttromagnetica). Le parti rilevanti sono state evidenziate con una linea blu laterale.
Scarica il materiale qui (cliccando sulla ruota dei colori):
L’ultimo aggiornamento delle regole di nomenclatura IUPAC* prevede delle variazioni in relazione alla nomenclatura degli alchini rispetto alle regole antecedenti, con particolare riferimento a quella che è la determinazione della catena principale.
Se il triplo legame (o se in generale i legami multipli) è contenuto nella catena più lunga, restano valide le regole presenti sul libro. Se, invece, esiste una catena più lunga che non è quella che contiene il triplo legame, bisogna fare attenzione. A differenza di quanto accadeva con le regole precedenti, ora la lunghezza della catena è più importante rispetto alla presenza del triplo legame (per cui al punto 3 pag. 292 il terzo esempio non segue le regole aggiornate).
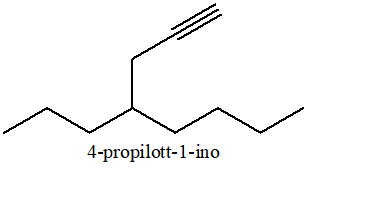
Ad esempio, il composto che segue, si chiama 4-etinilnonano: la catena più lunga è a 9 atomi di carbonio e non contiene il triplo legame. La porzione della molecola che contiene il triplo legame è dunque un sostituente, per il quale si usa il suffisso “-inil” (per indicare con “-in-” anche la presenza del triplo legame).
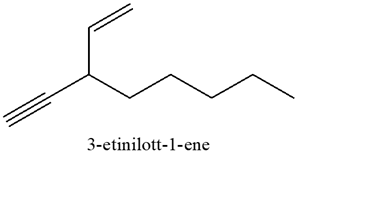
Quando necessario (cioè per sostituenti lunghi più di 2 atomi di carbonio) è necessario indicare anche la posizione del triplo legame, come nell’esempio seguente:
Se sono, invece, presenti due catene della stessa lunghezza, la catena principale sarà quella che contiene il triplo legame (e si deve quindi numerare la catena in modo da dare il numero più basso al primo carbonio sp). Esempio:
Se però una catena contiene il doppio e l’altra il triplo legame, a parità di lunghezza si sceglie come catena principale quella che contiene il doppio legame**:
Se ci sono più catene della stessa lunghezzacon legami multipli, si sceglie quella che contiene più legami multipli.
Di seguito sono mostrate le 5 catene della stessa lunghezza utilizzando colori diversi. Quella da scegliere è quella riportata in rosso (3 legami multipli vs. 1 e 2)
Si ricorda, infine, che in base all’ultimo aggiornamento IUPAC, il ciclo ha la precedenza sulle catene lineari (se non sono presenti gruppi funzionali). I legami multipli non modificano quest’ordine di priorità, per cui tutti i composti indicati sotto sono dei cicloesani sostituiti ai fini della nomenclatura (vedi i nomi indicati):
** Ricordiamo che se il doppio e il triplo legame sono presenti entrambi nella catena principale, restano valide le regole riportate sul libro: la catena va numerata in modo da dare il numero più basso possibile al primo legame multiplo che si incontra (sia esso il doppio o il triplo legame). Solo in caso di “parità”, il doppio legame avrà la precedenza rispetto al triplo, per cui si dovrà numerare nella direzione che dà il numero più basso possibile al primo carbonio sp2 che si incontra. Vedi esempi che seguono
Quando è necessario prevedere il meccanismo e il/i prodotti principali di una reazione dobbiamo porre attenzione, dove appropriato, alla regioselettività, stereoselettività e stereospecificitàdella reazione stessa. È dunque chiaro che lo studio preliminare dei meccanismi di reazione è essenziale. Nel rivedere tali meccanismi, provare a razionalizzare ogni passaggio in termini di reazione del nucleofilo con l’elettrofilo.
Nel descrivere il meccanismo di reazione, è importante fare un uso corretto delle frecce ricurve, per mostrare il movimento degli elettroni, e delle frecce adatte ad indicare il passaggio da uno stadio all’altro della reazione. Prestare, inoltre, attenzione alla presenza di eventuali atomi carichi positivamente o negativamente.
Per reazioni che prevedono la formazione di un intermedio carbocationico, fare sempre attenzione alla possibilità di trasposizioni che possano portare alla formazione di un carbocatione più stabile.
Seguire l’aspetto stereochimico delle reazioni
Per le reazioni stereospecifiche* (e, di conseguenza, stereoselettive), è necessario seguire la stereochimica della reazione utilizzando le opportune rappresentazioni grafiche (per i composti a catena aperta, usare strutture a cavalletto o formule prospettiche). Di seguito, due esempi.
Nel primo, facciamo reagire il (2E)-3-metil-2-pentene con Br2 in H2O. Mostriamo la stereochimica della reazione utilizzando le formule prospettiche.
Ricordiamo che il doppio legame è planare (i due carboni sp2 e gli atomi ad essi direttamente legati giacciono tutti in un piano); nell’immagine che segue, troverete i sostituenti legati ai carboni sp2 su cuneo pieno o su cuneo tratteggiato. Questo indica che stiamo immaginando che il piano su cui si trovano tutti i legami dei due carboni ibridati sp2 non è quello sello schermo (o del foglio), ma quello ad esso perpendicolare.
Gli elettroni π (pi-greco) potranno dare l’attacco all’elettrofilo sia al di sopra sia al di sotto del piano. Nell’immagine seguente è mostrato l’attacco verso l’alto. Si formerà quindi lo ione bromonio ciclico che subirà l’attacco del nucleofilo sul carbonio più sostituito. Questo attacco avviene in anti. Si formerà, quindi, un unico stereoisomero. (NB: per una descrizione dettagliata del meccanismo si rimanda al libro).
Da questa reazione stereospecifica (e quindi anche stereoselettiva) otterremo anche l’enantiomero del prodotto appena formato. Questo deriva dalla formazione dello ione bromonio sulla faccia inferiore del piano definito dai due carboni sp2.
NB: il prodotto ottenuto a seguito dell’attacco dell’acqua su questo ione bromonio avrà configurazione (2S,3R)
È possibile mostrare la stereochimica della reazione anche utilizzando le rappresentazioni a cavalletto. A titolo di esempio, facciamo avvenire la reazione sull’alchene (2Z)-3-metil-2-pentene (ci aspettiamo, dunque, la formazione di due composti che saranno tra loro enantiomeri, e saranno diastereoisomeri dei prodotti della reazione precedente). Per usare le strutture a cavalletto, riportiamo il doppio legame come mostrato di seguito (attenzione a rispettare la geometria del doppio legame!). Anche in questo caso, per attacco da parte degli elettroni π all’elettrofilo da sopra o da sotto al piano di formano due ioni bromonio. Qui è mostrato l’attacco verso il basso che porterà alla sintesi dello stereoisomero mostrato.
Per definire la stereochimica è consigliabile riportare la struttura su una proiezione di Fischer (ricordandosi di eclissarla prima di farlo).
A seguito della formazione dell’altro ione bromonio (che deriva dall’attacco al di sopra del piano), si otterrà l’enantiomero del composto qui ottenuto.
NB: avendo familiarità con le varie rappresentazioni delle molecole organiche, è possibile passare agevolmente dall’una all’altra. A questo punto potremmo, ad esempio, scrivere questo composto usando una struttura a segmenti.
La tabella 6.1 del libro può essere utile per verificare se la stereochimica della reazione è stata determinata correttamente
*Tutte le reazioni stereospecifiche sono anche stereoselettive (mentre non è sempre vero il contrario)
Analisi retrosintetica
Se per le reazioni stereospecifiche è necessario effettuare un’analisi retrosintetica (analisi necessaria per capire quale è l’alchene di partenza), è essenziale tener conto proprio della stereochimica con cui procede la reazione. È quindi fondamentale capire da quale alchene partire: per farlo, bisogna aver ben chiaro se la reazione prevede una stereochimica sin o anti.
Vediamo che succede se la reazione procede con stereochimica anti
ES.1: Partendo dall’opportuno alchene, illustrare il meccanismo di reazione della sintesi del seguente composto. Indicare eventuali altri prodotti fornendo per tutti il nome IUPAC completo di stereochimica.
Si tratta di una reazione di un alchene (3-metil-2-pentene) con bromo in metanolo. Poichè questa è una reazione stereospecifica, è ESSENZIALE capire da quale isomero (E o Z) dell’alchene bisogna partire per ottenere il prodotto desiderato. Dal momento che questa reazione è anche stereoselettiva, determiniamo le configurazioni assolute dei carboni chirali, sapendo che in questo caso otterremo anche l’enantiomero.
Per capire da quale alchene dobbiamo partire, trasformiamo questa proiezione di Fischer in una rappresentazione a cavalletto, sapendo che i sostituenti sulla linea verticale si trovano lontani dall’osservatore, mentre quelli sulla linea orizzontale sono rivolti verso l’osservatore:
L’addizione di bromo in metanolo procede con stereochimica anti; questo significa che Br e OMe devono trovarsi da parte opposta. Ruotiamo dunque la rappresentazione a cavalletto lungo il legame C2-C3 in modo da evidenziare quanto appena detto (dobbiamo ottenere il conformero sfalsato qui mostrato):
L’alchene di partenza è dunque (2Z)-3-metil-2-pentene (se la struttura a cavalletto è orientata come sopra mostrato e i sostituenti sono correttamente posizionati, basta a questo punto “eliminare” i due sostituenti e “aggiungere” il doppio legame come mostrato in figura).
Il seguente video può essere utile per visualizzare l’attacco in anti:
Vediamo che succede se la reazione procede con stereochimica sin
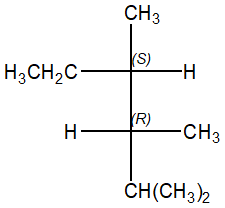
ES. 2: Da quale 2,3,4-trimetil-3-esene è possibile ottenere il seguente prodotto di idrogenazione catalitica?
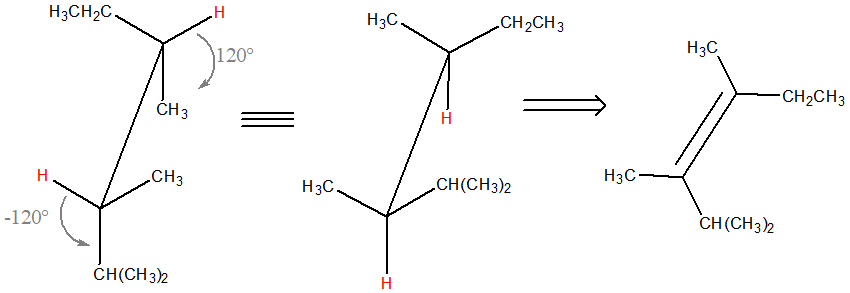
L’idrogenazione catalitica porta all’addizione di un idrogeno a ciascun carbonio sp2 e procede con stereochimica sin. La reazione è stereospecifica. Per capire da quale alchene dobbiamo partire, trasformiamo la proiezione di Fischer in una rappresentazione a cavalletto
L’addizione avviene con stereochimica sin; questo significa che i due H devono trovarsi dallo stesso lato. Ruotiamo dunque la rappresentazione a cavalletto lungo il legame C3-C4 in modo da evidenziare quanto appena detto (dobbiamo ottenere il conformero eclissato qui mostrato):
L’alchene di partenza è dunque (3Z)-2,3,4-trimetil-3-esene (se la struttura a cavalletto è orientata come sopra e i sostituenti sono correttamente posizionati, per definire l’alchene di partenza, basta a questo punto “eliminare” i due atomi di idrogeno e “aggiungere” il doppio legame come mostrato in figura). Da questo alchene, per idrogenazione catalitica, otterremo anche l’enantiomero del composto iniziale.
NB: in molti esercizi, una volta individuato l’alchene di partenza, viene poi richiesto di mostrare anche il meccanismo che porta alla formazione dei prodotti oppure viene chiesto di indicare eventuali altri prodotti (LEGGERE SEMPRE BENE LA TRACCIA).
Schemi riassuntivi e materiale didattico integrativo
Qui è possibile scaricare una tabella riassuntiva delle reazioni degli alcheni che può integrare lo schema che trovate sul libro alla fine del capitolo dedicato proprio a queste reazioni.
NB: per gli aspetti regiochimici delle reazioni per cui le informazioni non sono presenti nella tabella, far riferimento al testo e agli appunti delle lezioni (in quanto l’argomento richiede una trattazione più dettagliata; ad es.: la reazione che porta alla sintesi di aloidrine dà una regioselettività Markovnikov che è possibile capire analizzando il meccanismo della reazione).
Al link seguente (cliccare su “Learn more”) è possibile scaricare un secondo schema riassuntivo sulle reazioni degli alcheni studiate. Sono inoltre riportati, nello stesso file, alcuni approfondimenti relativi sia alle trasposizioni, sia agli orbitali coinvolti in alcune delle reazioni in questione:
Nel corso della reazione, si forma uno ione mercurinio a ponte come intermedio, che viene, a sua volta, attaccato dall’acqua. Questo stadio avviene con stereoselettività anti. In un secondo stadio, viene aggiunto NaBH4, ottenendo così il prodotto di riduzione. Questo passaggio rimescola la stereochimica, per cui il processo complessivo è un misto di addizione sin e anti.

































You must be logged in to post a comment.